Predicting quantitative traits from genome and phenome with near perfect accuracy
Abstract
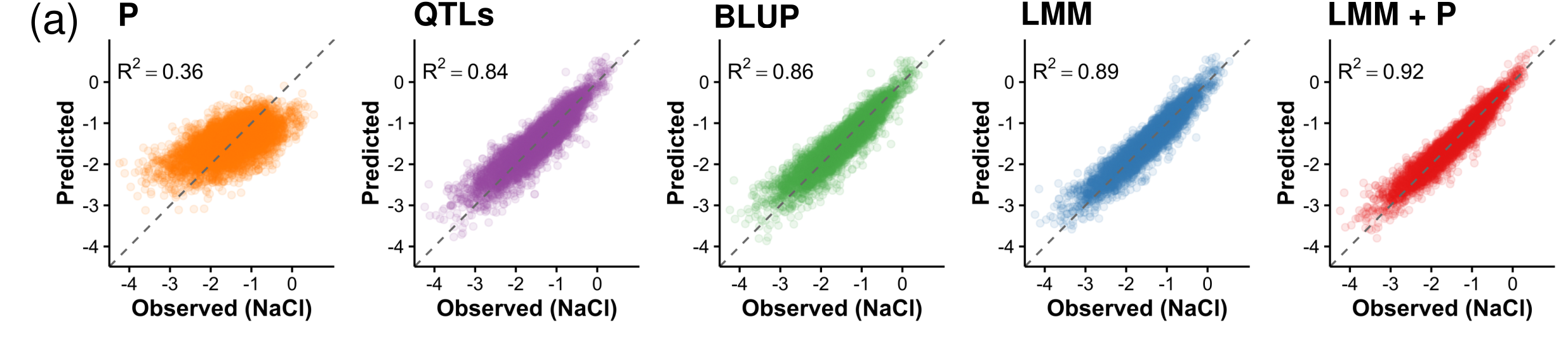
In spite of decades of linkage and association studies and its potential impact on human health, reliable prediction of an individual’s risk for heritable disease remains difficult. Large numbers of mapped loci do not explain substantial fractions of heritable variation, leaving an open question of whether accurate complex trait predictions can be achieved in practice. Here, we use a genome sequenced population of ∼7,000 yeast strains of high but varying relatedness, and predict growth traits from family information, effects of segregating genetic variants and growth in other environments with an average coefficient of determination of 0.91. This accuracy exceeds narrow-sense heritability, approaches limits imposed by measurement repeatability and is higher than achieved with a single assay in the laboratory. Our results prove that very accurate prediction of complex traits is possible, and suggest that additional data from families rather than reference cohorts may be more useful for this purpose.